Breakthrough in Long-Read Sequencing Unveils Extensive Human Genomic Diversity and Disease Links
July 23, 2025
Recent advancements in long-read sequencing (LRS) technologies have significantly enhanced the detection of structural variants (SVs) in human genomes, leading to the development of a nearly complete human pangenome reference.
A comprehensive study analyzed the genomes of 1,019 diverse individuals using LRS, uncovering a vast array of SVs that deepen our understanding of human genetic diversity and disease susceptibility.
Special focus was placed on the major histocompatibility complex, a key region for immune function, where variations have been linked to vaccine responses and autoimmune diseases.
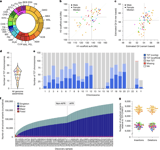
The dataset captured over 139,000 SVs, including deletions and insertions, with a majority being rare variants, highlighting the extensive genetic variation across populations.
The study was supported by multiple NIH grants and collaborations, underscoring the collective effort to advance genomic research.
Insights into the structure of survival motor neuron genes (SMN1/SMN2), associated with spinal muscular atrophy, were also gained, revealing potential disease-risk sites.
The research identified numerous inversions and segmental duplications, crucial for understanding structural polymorphisms in the human genome.
The study identified up to 26,115 structural variants per individual, totaling over 175,000 sequence-resolved events, illustrating the extensive structural diversity in human genomes.
Functional analysis showed that SVs can disrupt protein-coding genes, impacting nearly 1,000 genes and emphasizing their role in human genetics.
Genetic diversity was notably higher in African populations, with a median SV count per individual of nearly 24,000, compared to about 19,000 in other groups.
Researchers generated highly contiguous, haplotype-resolved assemblies for 130 genomes, closing 92% of gaps from previous references and achieving high-quality scores.
Analysis of centromere regions revealed remarkable diversity, with over 4,000 new variants identified among 1,246 centromeres, emphasizing their mutability.
A detailed classification of SVs uncovered extensive mobile elements and tandem repeats, vital for understanding human diversity and disease.
The study identified over 188,000 SVs, along with millions of indels and SNVs, representing a 59% increase in the SV callset compared to previous data.
Highly contiguous assemblies of the Y chromosome were achieved, providing new insights into its variability, despite the challenges posed by repetitive sequences.
The limitations of short-read sequencing in capturing certain SVs, especially insertions, were highlighted, emphasizing the importance of long-read data for comprehensive variation analysis.
A comprehensive dataset of SV alleles was created, revealing the distribution and origin of both common and rare variants across different populations.
The Human Genome Structural Variation Consortium analyzed genomes from 65 individuals worldwide, aiming to assemble nearly gapless chromosomes, including complex regions like centromeres.
Mobile element insertions constitute a significant portion of SVs, with full-length L1 insertions indicating active retrotransposition.
A new computational framework called SAGA was developed to improve SV discovery and genotyping through graph-aware analysis, enhancing classification accuracy.
Published in Nature, the study underscores how structural variations influence gene expression and contribute to understanding diseases and human adaptation.
This research enriches our genomic resources, advancing knowledge of human diversity and setting the stage for future genomics and personalized medicine studies.
Building on previous efforts by the Human Pangenome Reference Consortium, this study applied Oxford Nanopore Technologies to analyze SVs from the 1000 Genomes Project, covering diverse populations.
Using a combination of high-fidelity and ultra-long reads, researchers achieved about 99% completeness in genome assemblies, closing gaps in previous references.
Population-specific analysis revealed clear genetic differentiation, especially among African groups, highlighting the influence of ancestry on SV landscapes.
Summary based on 3 sources
Get a daily email with more Science stories
Sources
Nature • Jul 23, 2025
Complex genetic variation in nearly complete human genomes
Nature • Jul 23, 2025
Structural variation in 1,019 diverse humans based on long-read sequencing
Mirage News • Jul 23, 2025
Genetic Diversity Unveiled in Human Genomes Study